Applied Bioinformatics Course (ABC)
实用生物信息技术
2026-05-07更新
系统发生树构建课堂练习
课堂练习实例1 - 人的珠蛋白(Globiin)基因树构建
-
复制人的珠蛋白家族12个蛋白质序列
12 Globins
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment,
在数据类型Data Type弹出窗口中选择蛋白质Protein
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将12个珠蛋白序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有12条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果,注意所插入空位的特点
-
点击主菜单中数据Data按钮,在下拉菜单中选择系统发生分析Phylogenetic Analysis选项
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择邻接法Construct/Test Neighbor-Joining Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数改为100,
其它参数采用默认值
-
查看人的12个珠蛋白系统发生树,利用Subtree调整树的显示方式,和
实用生物信息技术课程教学实例
文中第四节图7进行比较,说明两者异同
课堂练习实例2 - 人、小鼠、大鼠珠蛋白系统发生树构建
-
复制人、小鼠、大鼠37个珠蛋白基因编码区CDS序列
37 Globins
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment,
在数据类型Data Type弹出窗口中选择DNA
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将37个珠蛋白编码区CDS序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW (Codons), 在弹出会话窗口中点击OK, 选择比对所有37条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果,注意所插入空位的特点
-
点击主菜单中数据Data按钮,在下拉菜单中选择系统发生分析Phylogenetic Analysis选项,在弹出会话窗口中点击Yes,
选择比对蛋白质编码序列Protein-Coding neucleiotide sequence data
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择邻接法Construct/Test Neighbor-Joining Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数改为100,
在替换类型Substitution Type中选择氨基酸Amino Acid, 其它参数采用默认值
-
查看人、小鼠、大鼠三个物种37个珠蛋白系统发生树,利用Subtree调整树的显示方式,和
实用生物信息技术课程教学实例
文中第四节图8进行比较,说明两者异同
-
查看人、小鼠、大鼠三个物种37个珠蛋白系统发生树,找出“先有物种、后有基因”和“先有基因、后有物种”的实例
课堂练习实例3 - 拟南芥和水稻SBP转录因子家族系统发生树构建
-
复制拟南芥和水稻36个SPL转录因子全长蛋白质序列
36 AtOsSPL
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment,
在数据类型Data Type弹出窗口中选择Protein
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将36个序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有36条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果
-
删除DNA结合结构域保守区上游和下游序列,点击主菜单Data按钮,选择输出比对结果Export Alignment, 选择输出格式FASTA Format,保存到本地文件
36AtOsSPLD.FAS
-
关闭并重新打开MEGA软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment, 在弹出窗口中选择从文件导入序列Retrieve Sequences From a file,
将上述36AtOsSPLD.FAS导入
-
点击主菜单中序列比对按钮,数据Data按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有36条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果
-
在下拉菜单中选择系统发生分析Phylogenetic Analysis选项,在弹出会话窗口中点击Yes
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择最大似然法Construct/Test Maximum Likelyhood Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数改为100,
其它参数采用默认值
-
查看拟南芥和水稻36个SPL转录因子系统发生树,利用Subtree调整树的显示方式,和
Gene Paper
文中图3进行比较,说明两者异同
课堂练习实例4 - 7个代表性脊椎动物alpha血红蛋白系统发生树构建
-
参阅Evolution 101演化生物学教学网站
Evolution 101
[PDF]
查看7个代表性脊椎动物系统发生树
7 Vertebrates
-
从UniProt数据库中找到上述7个代表性脊椎动物alpha血红蛋白,下载FASTA格式序列
7HBA.FAS
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment, 在数据类型Data Type弹出窗口中选择蛋白质Protein
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将7个alpha血红蛋白序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有12条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果
-
点击主菜单中数据Data按钮,在下拉菜单中选择系统发生分析Phylogenetic Analysis
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择邻接法Construct/Test Neighbor-Joining Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数改为100,
其它参数采用默认值
-
分析所构建的系统发生树是否能反映7个脊椎动物间的系统发生关系
课堂练习实例5 - 7个代表性植物18S rRNA系统发生树构建
-
从NCBI RefSeq参考序列数据库中找到6个代表性植物18S rRNA序列,下载FASTA格式序列
18S rRNA
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment, 在数据类型Data Type弹出窗口中选择DNA
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将6个18S rRNA序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有7条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果
-
点击主菜单中数据Data按钮,在下拉菜单中选择系统发生分析Phylogenetic Analysis
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择邻接法Construct/Test Neighbor-Joining Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数设置为500, 其它参数采用默认值
-
查看所构建的系统发生树,利用Subtree调整树的显示方式
-
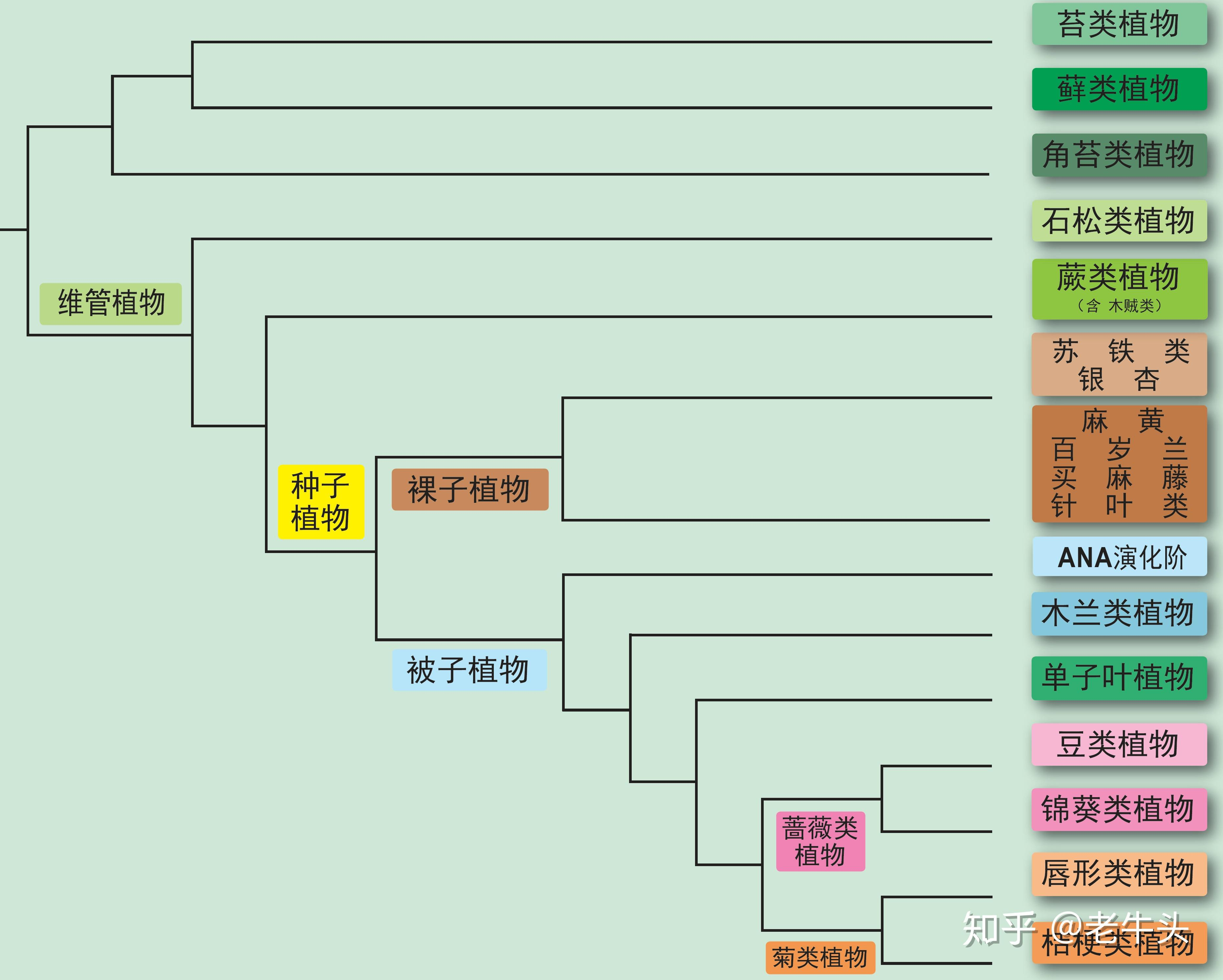
搜索植物分类网站,参考
绿色植物分类谱系
分析所构建的系统发生树能否反映6个代表性植物的系统发生关系
课堂练习实例6 - 非典病毒(SARS)变异位点DNA序列系统发生树构建
-
下载20个SARS病毒FASTA格式序列
20SARS
-
打开MEGA12系统发生树构建软件,点击主菜单中序列比对Align图标,在下拉菜单中选择创建Edit/Build Alignment,
在弹出会话窗口Alignment Editor中选择创建一个新的比对Creat a New Aligment, 在数据类型Data Type弹出窗口中选择DNA
-
删除空序列1. Sequence, 点击主菜单中编辑按钮Edit,在下拉菜单中选择粘贴Paste, 将20个SRAR病毒变异位点序列粘贴到编辑窗口中
-
点击主菜单中序列比对Alignment按钮,在下拉菜单中选择Align by ClustalW, 在弹出会话窗口中点击OK, 选择比对所有7条序列,
在ClustlW参数选择弹出窗口中点击OK,选择MEGA12给定的默认参数,查看比对结果
-
点击主菜单中数据Data按钮,在下拉菜单中选择系统发生分析Phylogenetic Analysis
-
在主菜单中点击系统发生Phylogeny按钮,在下拉菜单中选择邻接法Construct/Test Neighbor-Joining Tree,
在弹出窗口中将系统发生树稳定性测试Test of Phylogeny选项改为自举法Bootstrap Method, 将自举法重复次数设置为100, 其它参数采用默认值
-
分析所构建的系统发生树,阅读参考文献
SARS分子流行病学
[PDF]
推测非典病毒传播途径
{kind=link}